| Generate 2D atom coordinates gives overlapping atoms [message #1826] |
Tue, 31 January 2023 12:01  |
mcmc
Messages: 18
Registered: April 2018
|
Junior Member |
|
|
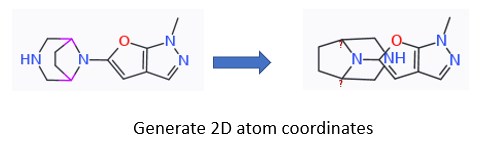
I have some bridged piperazines that get redrawn in a way that atoms are overlapping with the attached heterocycle upon generating 2D atom coords (which I do to superimpose the compounds in a set).
Hard to explain in words, but picture attached.
If these overlapping atoms could be avoided, that would be much appreciated.
|
|
|
|
| Re: Generate 2D atom coordinates gives overlapping atoms [message #1831 is a reply to message #1826] |
Thu, 02 February 2023 20:37  |
 nbehrnd
nbehrnd
Messages: 211
Registered: June 2019
|
Senior Member |
|
|
Hello mcmc,
your question does not describe how the piperazines on the left are generated in first place. Is it a SMILES string, a .sdf file you import? Do you draw them with DW's sketcher? Opting for the latter, starting to draw the molecule from the right, the furopyrazole, to which I added piperazine, and lastly, the bridge to piperazine, I did not observe the overlap you describe.
Regards,
Norwid
DW for Linux/MacOS version 5.5 with updates published by 2023-01-18 in Linux Debian 12/bookworm.
|
|
|
|
| Re: Generate 2D atom coordinates gives overlapping atoms [message #1835 is a reply to message #1831] |
Fri, 03 February 2023 10:51 |
mcmc
Messages: 18
Registered: April 2018
|
Junior Member |
|
|
Ah, no I didn't. They were either uploaded as SDF (V3000), or as SDF coming out of Chemdraw drawings.
I should also add, I modified the right-hand side from a different fused bicycle, for the purpose of this post. I did not want to disclose the actual molecules we are working on.
Also, the bridged piperazine is treated as substituent in generating the coords. The core (bicycle) is the constant part
I am surprised that this matters, as this module generates new 2D coords. But your observation is interesting, and I'll see if I can tune things a little. Maybe the size/depth of the bridge decides whether the 6- or 5-membered ring gets prioritised.
At any rate, it's a pity that overlapping atoms are generated in the first place, but maybe that's anyway not due to Datawarrior code.
[Updated on: Fri, 03 February 2023 10:57] Report message to a moderator |
|
|
|
| Re: Generate 2D atom coordinates gives overlapping atoms [message #1838 is a reply to message #1835] |
Fri, 03 February 2023 22:32 |
nbehrnd
Messages: 211
Registered: June 2019
|
Senior Member |
|
|
Dear mcmc,
I speculate an underlying contribution to the issue is the discrepancy between identifying a good orientation for a 3D object in space for a 2D projection, on one hand, and retaining a consistent pattern of single / double / triple bond lengths, on the other. A substituted briged piperazines may be an example where a chemist might consider to draw one single bond longer, than others to move and rearrange a (sub) motif on the paper/black board further away from the centre of the molecule to keep all well intelligible. On the other hand, a computer algorithm (like the one in DW, but equally in Marvin) may yield overlaps. The approach starting from 2D and a sketcher like ChemDraw/ChemDoodle is different as all work is in 2D only.
Because you mentioned the structure import into DW used .sdf, it was possible to identify COD 1513802 of the Crystallographic Open Database as an entry where DW faces a similar problem to the one reported by you. But it isn't unique to DW; the same structure eventually imported into Marvin equally has no orientation without a (partial) overlap of the atoms.
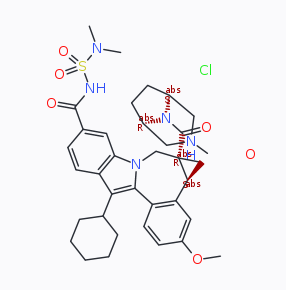
With regards,
Norwid
|
|
|
|
|
|
| Re: Generate 2D atom coordinates gives overlapping atoms [message #1849 is a reply to message #1848] |
Fri, 10 February 2023 23:00 |
nbehrnd
Messages: 211
Registered: June 2019
|
Senior Member |
|
|
Dear mcmc,
I do not recall if one can toggle on/off the stereochemical descriptors in DW's sketchers. If absent temporarily, the representation of the example from COD would be even lighter / less stuffed, than now (compared to the result by MarvinJS).
On the other hand, departing from a 3D structure imported by read of a .sdf, picking the best perspective to flatten the representation into 2D possibly stays a difficult task (option a, the example with MarvinJS). On the other hand, I speculate shredding the 3D information about coordinates to obtain a reduced representation as a string (e.g., SMILES, or DW's idnode) as an intermediate which subsequently is used to sketch the molecular structure from scratch may yield less overlaps (option b). Based on the comparison of the results, possibly DW takes this second route b.
For the COD compound, DW assigns CN(C)S(NC(c(cc1)cc2c1c(C1CCCCC1)c(-c(cc1)c([C@@H]3C4)cc1OC)[ n]2C[C@]34C(N1[C@@H]2C[NH](C)C[C@H]1CC2)=O)=O)(=O)=O.[O].[Cl ] as SMILES string. Depending if CIP labels are enabled, or not; and groups are abbreviated, or not (e.g. cyclohexyl as Cy), the implemented algorithm by CDK Depict yields
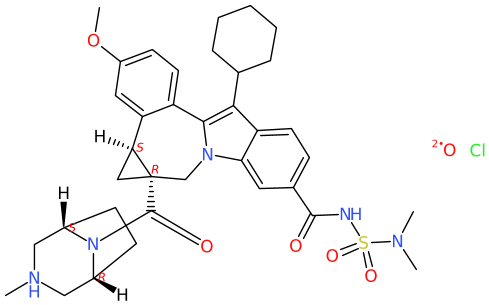
Obviously, the structure formula includes an overlap, though the absence of the wedges is indicative enough to recognize that this. Bond lengths are not uniform. The project's choice for «smart chiral Hydrogens» arguably keeps the inner of the cyclopropane moiety and bridged piperazine less crowed.
With regards,
Norwid
Web site: https://www.simolecule.com/cdkdepict/depict.html
source code: https://github.com/cdk/depict
|
|
|
|

 Search
Search Help
Help Members
Members Register
Register Login
Login Home
Home









")
