Home » DataWarrior » Functionality » Doubly Substituted Reactants for Combinatorial Library
| Doubly Substituted Reactants for Combinatorial Library [message #1315] |
Sun, 20 June 2021 00:32  |
 sn
sn
Messages: 4
Registered: June 2021
|
Junior Member |
|
|
Hello everyone,
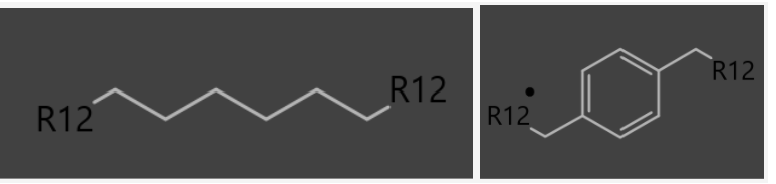
I'm trying to create an original reaction(Fig1) which has two substituents(R12) and a variety of linkages(Fig2). But I couldn’t define these linkages by “any atomic number”.
Compounds in Fig3 are only accepted series but limited to a single atom between substituents(R12).
In the case of a single substituent, “any atomic number” always works for a variety of substituents(Fig4,5).
Is there any way to overcome this problem?
Thank you very much for your help and let me know if you need any additional information.
Best Regards,
sn
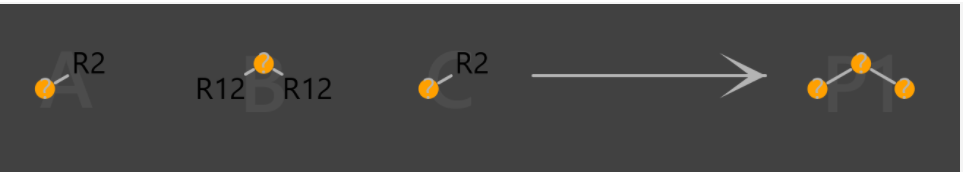
Fig1.
… so on.
Fig2.

Fig3.
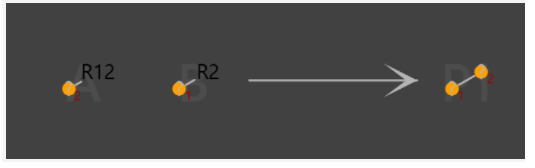
Fig4.
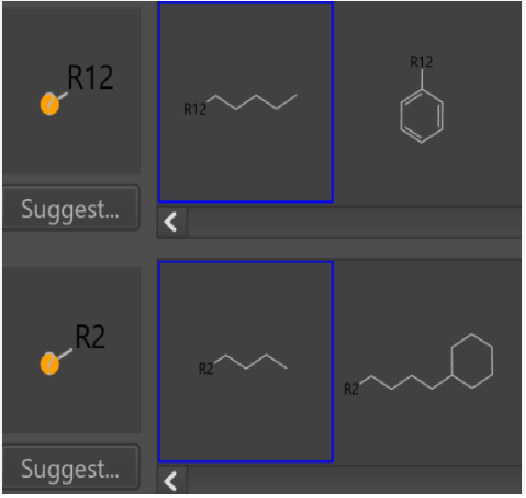
Fig5.
-
 Attachment: 5.png
Attachment: 5.png
(Size: 23.80KB, Downloaded 1256 times)
-
Attachment: 4.png
(Size: 8.39KB, Downloaded 1176 times)
-
Attachment: 3.png
(Size: 8.65KB, Downloaded 1161 times)
-
Attachment: 1.png
(Size: 17.90KB, Downloaded 1193 times)
-
Attachment: 2.png
(Size: 15.96KB, Downloaded 1091 times)
|
|
|
|
|
|
|
|
| Re: Doubly Substituted Reactants for Combinatorial Library [message #1322 is a reply to message #1321] |
Thu, 24 June 2021 06:46  |
nbehrnd
Messages: 241
Registered: June 2019
|
Senior Member |
|
|
Dear sn,
from the documentation and some limited testing with DW I infer that this (currently) is not yet possible this way. Perhaps a misunderstanding by mine, but I infer «any atom number» is about «at this position, the atom may be a carbon, a nitrogen, an oxygen, etc», rather than the number of atoms (like let there be one atom, or two, three, etc.). On the other hand, one may prepare a list of this e.g., diamines separated by one, two, three methylene groups in advance. Past mapping the reaction in the tab «generic reaction», the subsequent tab «reactants» then allows to add them from a e.g., from a .dwar, .sdf, .mol2 file.
As an example, a bis-substitution like defined by
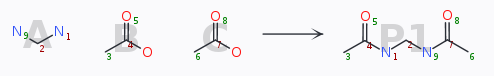
yields a list like
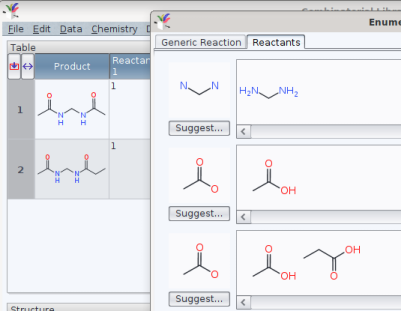
To obtain the special permutation «use the same acid on the left and on the right hand amine» would then justify to load and re-use such a pre-defined list to cover the chemical space efficiently.
Norwid
-
Attachment: setup.png
(Size: 4.74KB, Downloaded 1076 times)
-
Attachment: example.png
(Size: 28.09KB, Downloaded 1091 times)
|
|
|
|
|
|
| Re: Doubly Substituted Reactants for Combinatorial Library [message #1326 is a reply to message #1323] |
Fri, 25 June 2021 20:52 |
nbehrnd
Messages: 241
Registered: June 2019
|
Senior Member |
|
|
Dear SN,
you may consider chemical graph generators to generate list of reactants as .sdf files. Two examples:
OMG requires Java 1.8, e.g. openjdk version 1.8.0_242 (one may still find in Xubuntu 18.04 LTS), or java 8.0.292-zulu (however OpenJDK 11.0.11 e.g., in Linux Debian bullseye will (currently) not work). Then, an input like
java -jar OMG.jar -ec C4H12N2 -o out_bisamine.sdf
yields a list of hydrocarbons including those with two amines. Some of the entries (see .sdf below) may be «surprising» from a chemical point of view. However a) you may pre-define non-overlapping fragments which must be present to guide the generator. Remember that you may create an empty structure table, populate this first column with DW structure editor, and eventually export this as .sdf (File -> Save Special) and then run
java -jar OMG.jar -ec C4H12N2 -o out_bisamine.sdf -fr fragments.sdf
to constrain the generation. And b), once the list is imported into DW, use e.g. the entries below Chemistry -> From Chemical Structure -> Calculate Properties to remove suggestions by ring counts / heterocyclic ring count, H-donors / acceptors, etc.
For MOLGEN there is a dedicated test page. Note, here Hill formulae may be provided as range; this may fit e.g., your aim to define diamines with a varying number of methylene groups as spacer. Based on their example 2 about halogenated halogens, the input criterion C1-3N1-2H4-10 yields 468 suggestions with one or two amino groups. The generation may be constrained, e.g. skipping cycles (set cycles = 0), and double bonds (set maximal bond multiplicity = 1) as parameters in addition. Then, there are just 29 entries matching all three criteria. Or, derived from example 4, a sum formula of C[sp2_a]6H4-6N[val=5,d=2]0-2O0-4 altogether with the limit of one cycle only, yields a list of 74 molecules, including benzenes with one or two nitro groups in ortho/meta/para relationship.
The web site offers only a glimpse of the program. The possibility to define fragments required / fragments to skip which the web page not offers is not shown, but present in limited (Windows) versions of the program one may download. Equally, the web site's work stops the molecule generation with entry #1000. Despite their rough conformations, the suggestions displayed are good enough to be exported as .sdf to continue processing in DataWarrior (the filtering mentioned above): just open JSmol's pulldown menu, File -> Save -> Save a copy of genXYZ.sdf to fetch the information.
Below a selection of output .sdf generated by OMG / MOLGEN.
Norwid
[Updated on: Fri, 25 June 2021 20:57] Report message to a moderator |
|
|
|
Goto Forum:
Current Time: Sun Jul 26 07:35:13 CEST 2026
Total time taken to generate the page: 0.00586 seconds
|



